What is Fabry disease?
Fabry disease (also known as alpha-galactosidase A deficiency) is a rare genetic condition caused by the lack of an enzyme called alpha-galactosidase A. This enzyme usually breaks down a chemical called GL-3 and without it, the GL-3 builds up in various organs of the body. High levels of GL-3 in the kidneys can lead to kidney failure.
Fabry disease was first identified in 1898 by two British clinicians, Dr William Anderson and Dr Johannes Fabry, and is sometimes also known as Anderson-Fabry disease.
It affects around 1 in 40,000 people in the UK. It almost always only affects males.
What are the signs and symptoms of Fabry disease?
Common symptoms of Fabry disease include:
- Pain in the hands and feet. This is often worse after sudden changes in temperature and can also be triggered by tiredness, stress and exercise.
- Problems regulating body temperature, including an inability to sweat, which can lead to over-heating.
- Stomach pain, especially after eating.
- Frequent bowel movements.
- Streaks in the cornea of the eye that look like a starburst but don’t affect the vision. In some cases cataracts can develop in one or both eyes.
- Tinnitus (ringing in the ears).
- Extreme tiredness.
- Clusters of small reddish-purple dots on the skin around the belly button. These do not cause any pain and are not infectious.
Kidney function is usually normal in children but gets worse over time. Kidney failure usually occurs in early adulthood.
What causes Fabry disease?
Chemical changes in the body occur due to enzymes, which act like parts of a machine in the body’s factory. Fabry disease occurs when not enough of an enzyme called alpha-galactosidase is produced, or none is produced at all. This enzyme normally breaks down a type of fat called GL-3. Without it, GL-3 builds up and affects the functioning of the body’s organs.
How is Fabry disease diagnosed?
Fabry disease is usually diagnosed by a blood test that examines levels of GL-3.
The main symptoms of pain in the hands and feet, an ability to sweat, and the presence of reddish-purple spots on the stomach are key indicators of the condition.
Urine tests are used to check for signs of kidney damage and genetic testing may also be offered.
Does Fabry disease affect other parts of the body?
GL-3 can build up in the heart as well as in the kidneys. This can affect how well the heart contracts to pump blood, disrupts the nerves in the heart and makes the heart grow extra-large. All of these can cause an irregular heartbeat and affect the functioning of the heart’s valves, which increases the risk of heart attacks.
GL-3 can also build up in the brain, narrowing or blocking the blood vessels and potentially leading to a stroke or mini-stroke (TIA).
Does Fabry disease run in families?
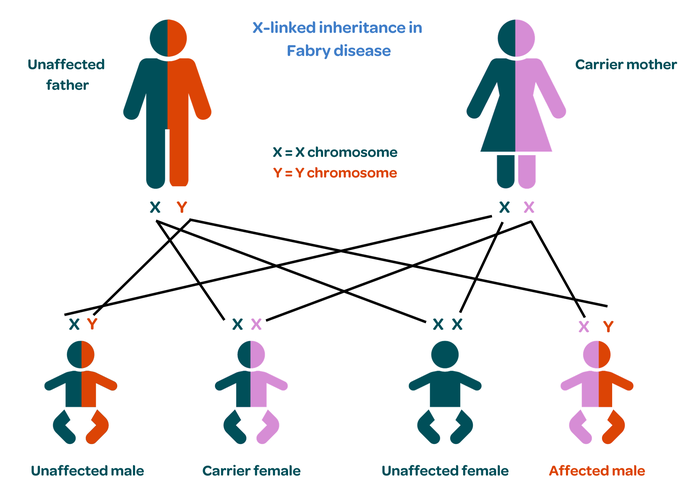
Fabry disease is a genetic condition so it can be inherited. It is caused by an abnormality or mutation in a gene called GLA, which is found on the X-chromosome.
Women have two X chromosomes, so the healthy gene compensates for the damaged one. However, men only have one X chromosome, so they have no back up if the gene is damaged. This is why Fabry disease occurs almost exclusively in men, although a very small number of cases have been diagnosed in women. These are usually mild with few symptoms.
Women with one damaged gene are known as carriers. There is a one in two chance that they will pass the damaged gene onto their children. If they pass it onto their daughter, she will also be a carrier. If they pass the gene onto their son, he will develop the condition.
Affected men cannot pass the condition onto their sons as they inherit the Y chromosome but will pass it onto their daughters who will be carriers.
If a parent has been diagnosed with Fabry disease or is a confirmed carrier, pre-natal and/or pre-implantation genetic diagnosis (PGD) may be offered. This involves testing embryos for Fabry disease during a cycle of in vitro fertilisation (IVF) and only transferring unaffected embryos to the mother’s womb to ensure the birth of a child without the condition.
How is Fabry disease treated?
Recent developments in enzyme replacement therapy have been shown to be very effective in treating Fabry disease.
Injections of the enzyme alpha-galactosidase remove GL-3 from the blood and tissue where it has built up. If treatment is started in early childhood, the complications normally seen with Fabry disease are unlikely to occur and people can lead normal lives.
If treatment is started later, any damage that has already occurred will not be reversed, but it will not get any worse. Injections are needed every two weeks. The first few doses are given in hospital to monitor for any side effects, but treatment can then continue at home. The treatment is currently available for people aged over seven in the UK.
Chaperone therapy (migalastat) may also be offered for adults with Fabry disease, depending on the precise genetic change that causes the condition. Chaperone therapy stabilises the damaged alpha-galactosidase enzyme and allows it to work properly. It is given as a tablet every other day. It is currently only approved for use in adults in the UK.
If kidney failure occurs, dialysis and/or transplant may be needed. The new kidney is protected from the effects of Fabry disease as it contains a small amount of the alpha-galactosidase enzyme. However, problems can still re-occur in other parts of the body.
Where can I get more information or support about Fabry disease?
For more information on Fabry disease, including its diagnosis, symptoms and treatment, visit the MPS Society.
Publication date: 11/2023
Review date: 11/2026